Learn how our work, shaped and funded by the public, saves lives and improves the quality of life of patients, service users, carers and communities.
These are the stories of the impact of our research on health, science and the UK economy.
Read our full list of stories
Latest stories

A clinical scoring test developed by NIHR-funded researchers has cut the use of antibiotics prescribed for sore throats by nearly a third and a cost-effective approach to manage patients’ symptoms.

Use of new software developed with NIHR funding is identifying more patients at high risk of early heart disease and heart attacks. Earlier diagnosis and treatment are saving lives and shaping healthcare policy.

In the biggest cystic fibrosis trial in the UK, NIHR-funded research has shown that a personalised web platform, CFHealthHub, could markedly increase adherence to treatment.

An NIHR-funded evaluation showed referring people with pre-diabetes to the NHS Diabetes Prevention Programme cut their risk of progressing to diabetes by 20%.

The CAP-IT trial of antibiotic use in young children with pneumonia has delivered practice-changing results, showing the length of treatment can be reduced.

An NIHR Clinician Scientist award supported Professor Angus Jones and his colleagues in developing convenient tests to confirm patients’ diabetes diagnosis.
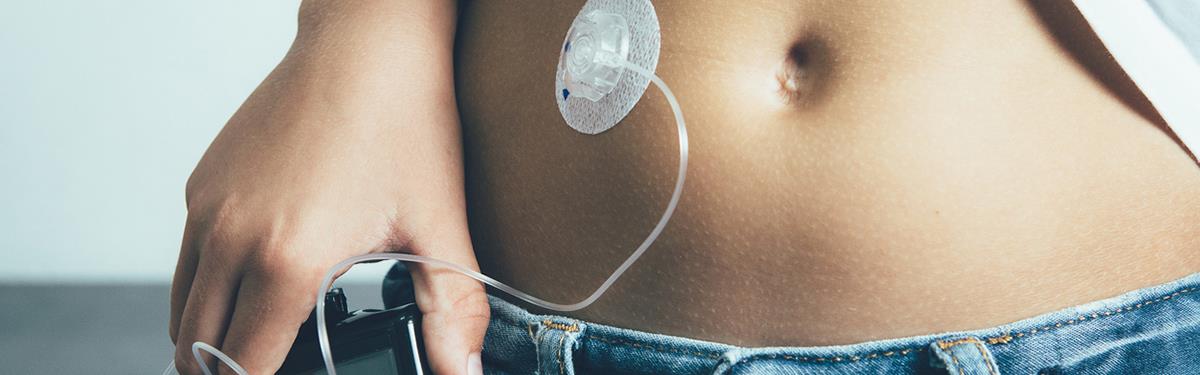
An artificial pancreas developed by NIHR-supported researchers and recommended by NICE is changing the lives of people with type 1 diabetes.

Researchers are changing the course of continence care for people living with dementia in hospital after finding usual care cultures promote incontinence.

Researchers found that a high-sensitivity blood test ruled out heart attacks in patients with chest pain, allowing many to be discharged from hospital.

Researchers have shown that a simple, low-cost change of gloves and instruments during surgery can reduce life-threatening surgical site infections.
